![]()
Tutorial: Parameterization of Metalloproteins
Overview
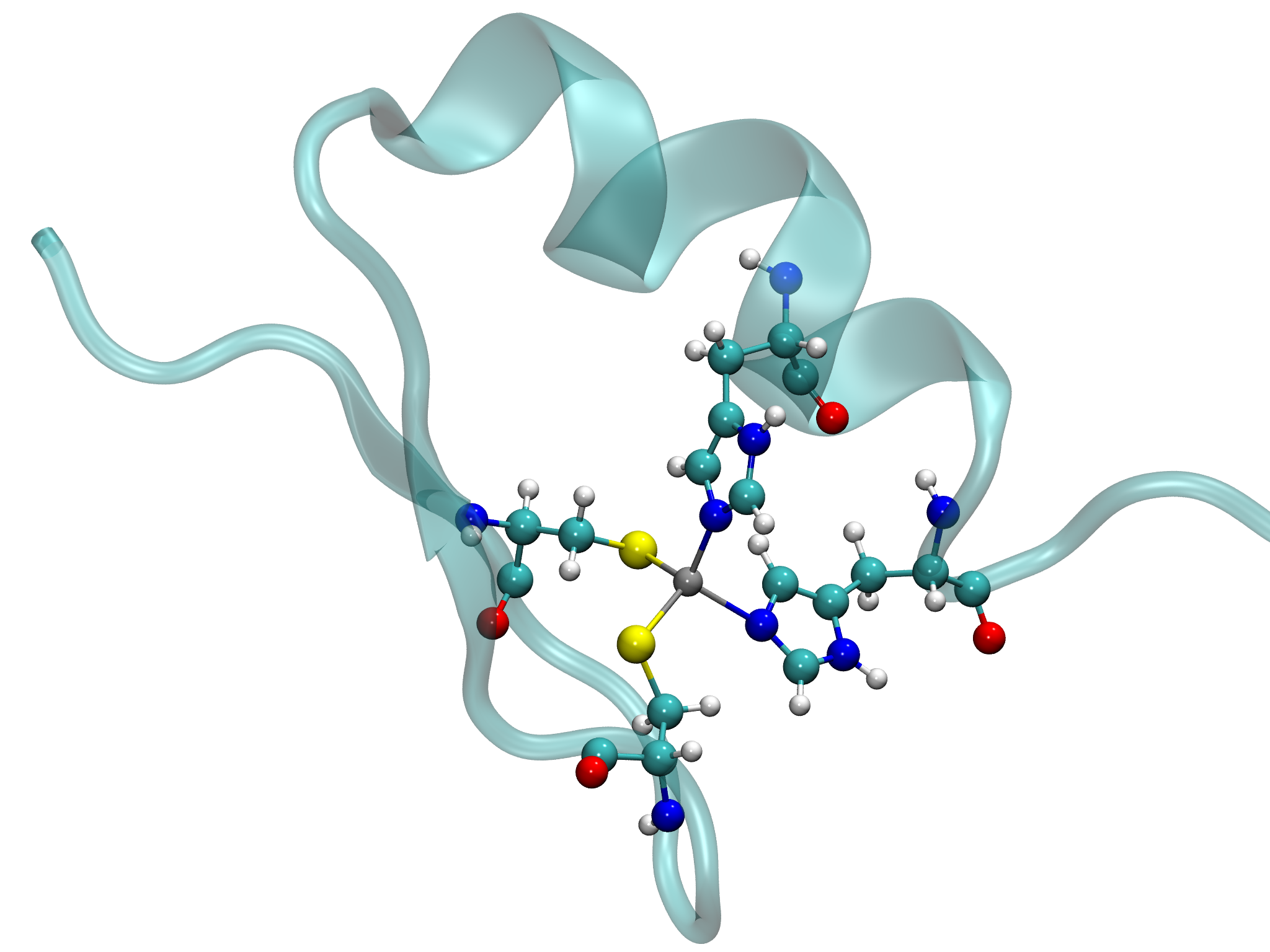
This tutorial provides a step-by-step guide for parameterizing a metalloprotein using Gaussian for QM calculations. The same workflow can be applied to ORCA and GAMESS as well. The example used throughout the tutorial is 1SP2.pdb, a protein containing a zinc atom coordinated to two cysteines and two histidines amino acids, as shown in the figure below.
1. Preprocessing the PDB File
After downloading the PDB file from the Protein Data Bank, preprocessing is necessary to ensure compatibility with easyPARM and molecular dynamics (MD) simulations.
1.1 Preprocessing Tools
You can use one of the following tools:
Important Notes:
- H++ may remove the metal ion or non-standard residues. After using H++, manually add the missing metal and its linked non-standard residues if necessary.
- Protonation states of histidine and cysteine may not be correct after preprocessing. Verify the PDB file and make the necessary adjustments: cysteine is labeled as CYS but should be CYM, and the same applies to histidine.
- Protonated form: protonated.pdb
- Deprotonated form: deprotonated.pdb
- Correct Orientation of Side Chains: Check the orientation of ASN, GLN, and HIS groups. The process of correcting orientations and adding hydrogens can sometimes shift the positions of residues linked to the metal. Verify that the metal and its coordinating residues are positioned
- Ligands (Non-standard Residues): Ensure all ligand atoms have unique names in the PDB file. Tools such as reduce (from AmberTools), Avogadro, or GaussianView can be used to add hydrogens if missing. Example correction for duplicate atom names:
Incorrect: ATOM 528 H LIG A 31 -12.251 -8.336 -3.849 1.00 9.27 H ATOM 529 H LIG A 31 -11.251 -7.336 -2.849 1.00 9.27 H Corrected: ATOM 528 H1 LIG A 31 -12.251 -8.336 -3.849 1.00 9.27 H ATOM 529 H2 LIG A 31 -11.251 -7.336 -2.849 1.00 9.27 H - Ensure no missing atoms in the structure.
Once the structure is corrected, save the final PDB file:
- Final metalloprotein PDB: final_complex.pdb
2. Generating the XYZ Structure with easyPARM
Run easyPARM and select option 2:
Select your option:
1- Generate molecular complex parameters
2- Generate metalloprotein .xyz structure
3- Convert AMBER parameters to OpenMM or GROMACS format
Enter your choice: 2
Please provide the metalloprotein pdb file: final_complex.pdb
XYZ Output: initial_structure.xyz
This step prepares the XYZ file initial_structure.xyz required for:
- Optimization
- Frequency calculations
- Charge calculations
3. QM calculation Outputs
- Optimized structure: OPTIMIZED.xyz
- Frequency calculation log: OPT_FREQ.log
- Charge calculation log: CHARGES.log
- Formatted checkpoint file: COMPLEX.fchk
4. Generating Molecular Parameters with easyPARM
You can run using the non-interactive mode using the template for the input in non-interactive section, or you can proceed with the interactive mode as described below.
Run easyPARM again and select option 1:
Enter your choice: 1
This will prompt a series of configurations:
4.1 AMBER Configuration
Choose how to load AMBER:
- Option 1: Use currently loaded AMBER
- Option 2: Specify AMBER installation path
Since AMBER is already loaded, select option 1:
Enter your choice: 1
4.2 System Charge
The tool will request the total charge of the system:
Please provide the total charge: 0
4.3 Providing the Optimized Structure for the Seminario Method
Specify the optimized XYZ geometry file:
Please provide the optimized XYZ geometry file: OPTIMIZED.xyz
4.4 Charge Calculation Method
Choose a method from the menu:
Select the charge calculation method:
1- GAUSSIAN (RESP charges)
2- ORCA (CHELPG charges)
3- ORCA (RESP Charges)
4- GAMESS (RESP Charges)
5- GAMESS (GAMESS Fit Charges)
Enter your choice: 1
4.5 mol2 File Generation
Select the charge output format:
Please select the input format:
1- Gaussian Output (.log file)
2- RESP (.gesp file)
Enter your choice: 1
Select the charge method:
Please select the charge method (recommended: RESP):
1- RESP (resp)
2- Mulliken (mul)
3- ESP (esp)
4- AM1-BCC (bcc)
Enter your choice: 1
Select the atom type:
Please select the atom type:
1- AMBER Force Field (AMBER)
2- General AMBER Force Field (GAFF)
3- General AMBER Force Field (GAFF2)
Enter your choice: 2
Provide the charge output file:
Please provide the charge output file (e.g., .log, .gesp): CHARGES.log
4.6 Seminario Method Setup
Select the format for the Seminario method:
Please select the format you will provide:
1- Orca Output
2- Gaussian Output
3- Gaussian Checkpoint
4- Gaussian Formatted Checkpoint
5- Gamess Output
Enter your choice: 4
4.7 Metalloprotein Structure Confirmation
Does your structure belong to MetalloProtein? (y/n): y
Provide the final metalloprotein PDB file:
Please provide the metalloprotein PDB file: final_complex.pdb
4.8 Change Residue ID
Would you like to change the residue ID (Default= mol)? (y/n): n
5. Output Generation
Upon completion, easyPARM generates the following files:
5.1 Key Outputs
- Mol2 files (Residues & Metal):
- Force Field Parameters: COMPLEX.frcmod
- Metalloprotein PDB: easyPARM_MetalloProtein.pdb
- Bond Information: Bond_Info.dat
- New Atom Type Definitions: Hybridization_Info.dat
5.2 Explanation of Output Files
Mol2files contain molecular structures of residues and metal.COMPLEX.frcmodcontains the derived force field parameters.easyPARM_MetalloProtein.pdbis the final protein structure including the metal, for use in tleap.Hybridization_Info.datdefines new atom types and hybridization states.Bond_Info.datlists metal-residue bonds, which must be manually added in tleap.
6. Atomic Charge Restraint (Optional)
The tool allows you to restrain atomic charges if needed. In this case, select No:
Would you like to restrain the charge on specific atoms? (Yes or No): N
7. Running tleap
Once the parameters are generated, you can proceed with tleap to prepare the final topology and coordinate files. Use the appropriate tleap input file to ensure correct setup:
Streamlined Metalloprotein Library Integration
Our latest implementation now supports the automatic generation of a library file for metalloproteins, eliminating the need for manual handling of multiple parameter files. The generated COMPLEX.lib file simplifies the tleap process by allowing direct loading with:
loadoff COMPLEX.lib
With this approach:
- There is no need to use the
Hybridization_Info.datfile. - You do not need to load each individual
.mol2file manually. - The metalloprotein setup is now more efficient and automated.
tleap Input File
Use the following tleap input file for reference:
- tleap input file: tleap.in
This completes the parameterization of the metalloprotein.