![]()
Tutorial: Parameterization of Multi-metal Metalloproteins
Overview
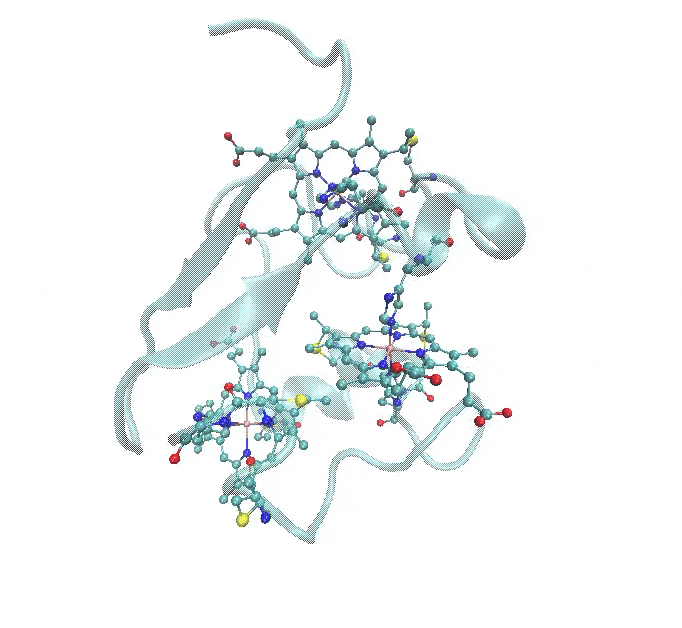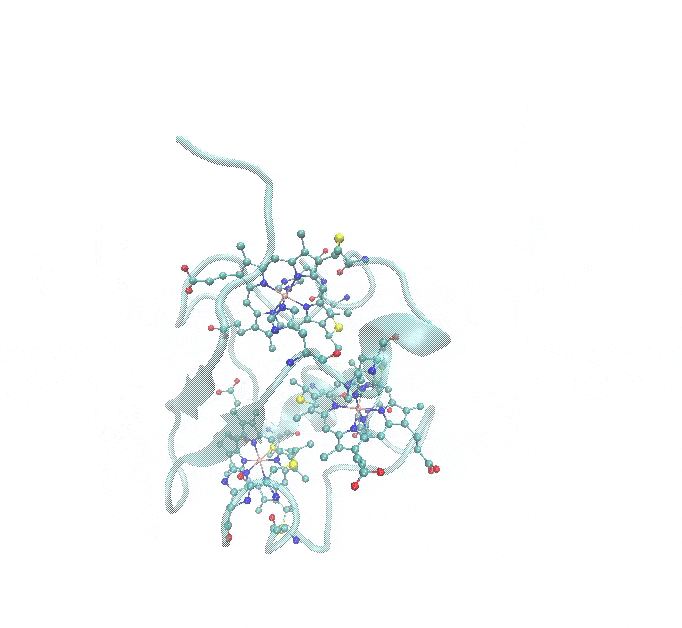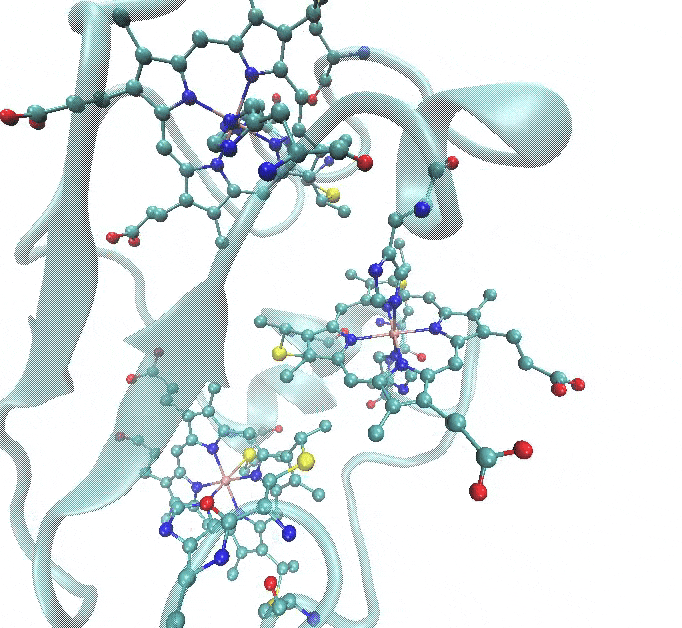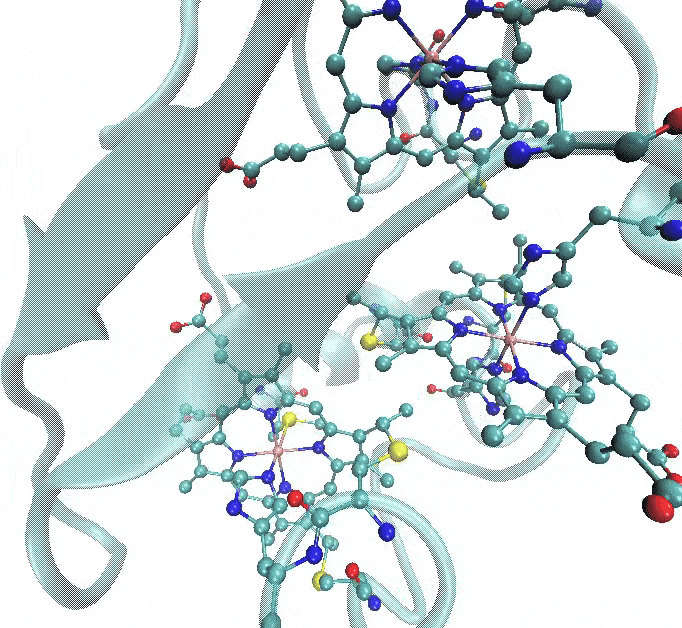
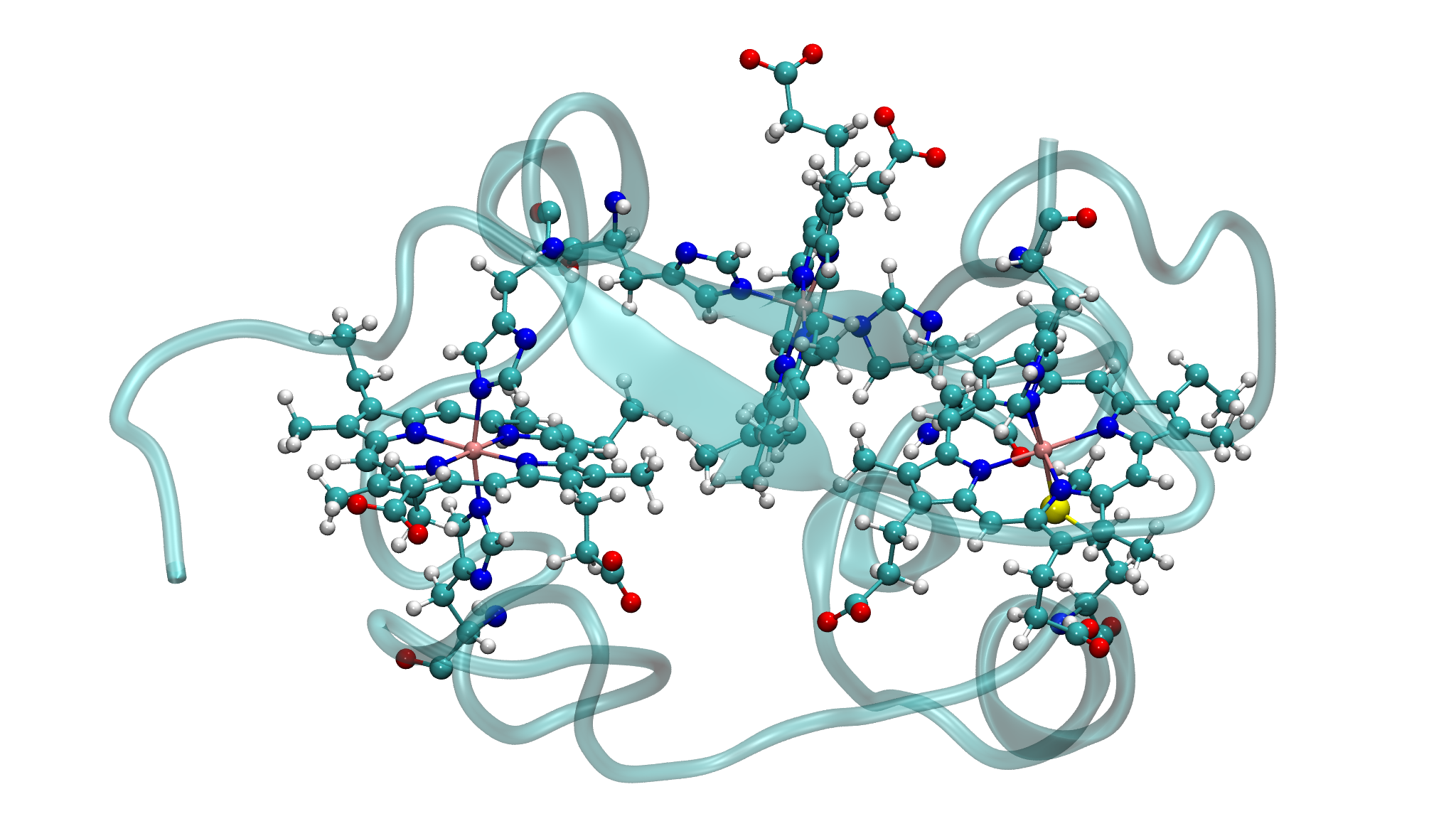
This tutorial provides a step-by-step guide for parameterizing a multi-metal metalloprotein using Gaussian for QM calculations. The same workflow can be applied when using ORCA and GAMESS as quantum-chemistry software. The example used throughout the tutorial is 1RWJ.pdb, a protein containing a three-heme c structure, each containing an iron (Fe) atom, as shown in the figure below.
Parameterization Challenges and Strategy
This tutorial highlights the capabilities of easyPARM by demonstrating how to parameterize a complex metalloprotein structure. The main challenges include:
- Multi-Metal System: The protein contains three heme c groups, each with an Fe atom that needs parameterization.
- Ligand Coordination Complexity:
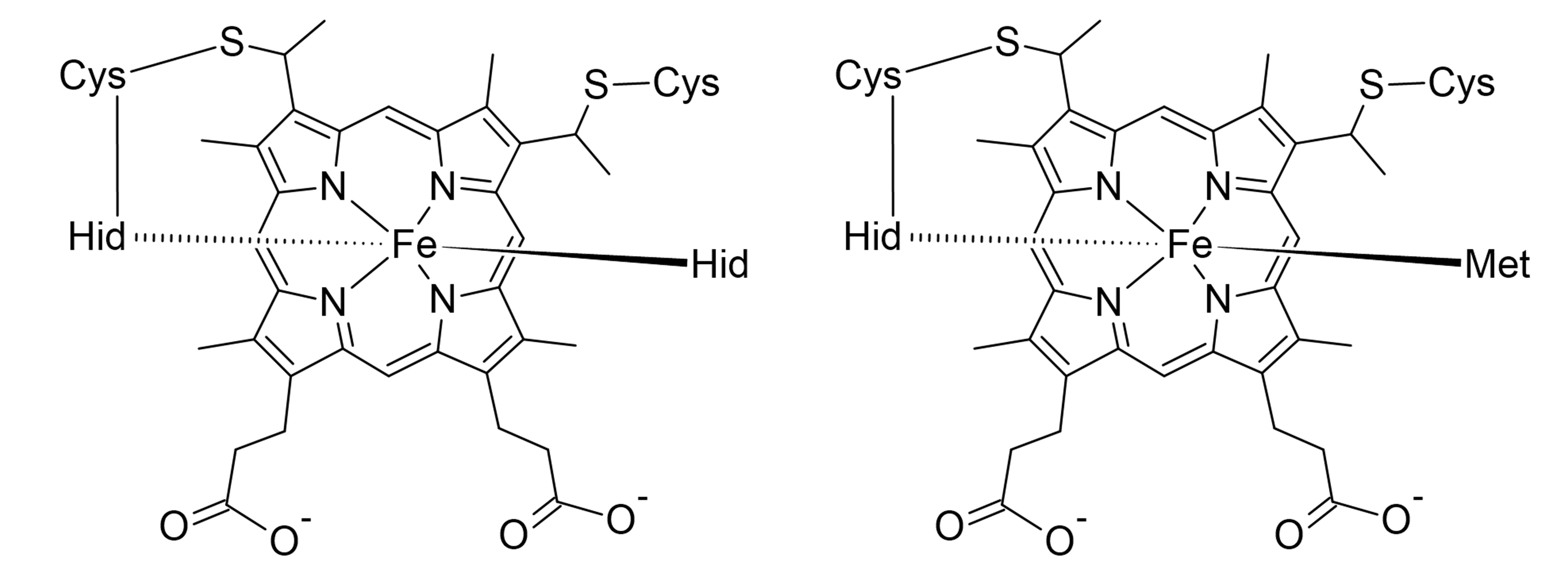
- Two of the heme c groups have Fe coordinated to two histidines (HID), and the porphyrin part is linked to the protein through a thioether bond with two cysteines (CYS). This means that parameterization is required not only for the metal-protein interactions but also for the non-standard thioether linkage.
- The third heme c group has a slightly different coordination, where one of the histidines is replaced by a methionine (MET).
The figure below illustrates these coordination environments:
This workflow ensures accurate force field parameters for multi-metal systems, making it easier to simulate metalloproteins with complex coordination environments.
1. Preprocessing the PDB File
After downloading the PDB file from the Protein Data Bank, preprocessing is necessary to ensure compatibility with easyPARM and molecular dynamics (MD) simulations.
1.1 Preprocessing Tools
We will use the following tool:
- H++ to prepare the system 1RWJ.pdb. After using H++, you will notice that the metal and its non-standard residues are removed, leaving only standard residues in the output file prepared.pdb.
Important Notes:
- Protonation states of residues linked to the metal, such as histidine and cysteine, may not be correct after preprocessing. Verify the PDB file and make necessary adjustments:
- Cysteine (CYS) should be renamed CYM, with the hydrogen linked to sulfur removed.
- Histidine (HIE) should be renamed HID, with the hydrogen linked to nitrogen removed.
- Protonated form: protonated.pdb
- Deprotonated form: deprotonated.pdb
1.2 Manual Preparation
Once you have the deprotonated PDB, you only need to extract the non-standard residues to add the missing hydrogens. In this case, the non-standard residues correspond to the three heme groups (HEC) from 1RWJ.pdb, corresponding to residues 90, 91, and 92. This procedure follows case 3
Since hydrogen atoms are missing in these structures, use reduce from AmberTools to add them:
reduce -i ligand1.pdb -o ligand1_h.pdb
reduce -i ligand2.pdb -o ligand2_h.pdb
reduce -i ligand3.pdb -o ligand3_h.pdb
Then, create three copies of deprotonated.pdb as:
- protein1.pdb
- protein2.pdb
- protein3.pdb
Each ligand must be added to its corresponding protein file individually. Follow these steps to create three separate protein-ligand complexes:
Ensure you have the following files ready:
ligand1_h.pdb, ligand2_h.pdb, and ligand3_h.pdb.
Add ligands to the corresponding protein files
ligand1_h.pdb → protein1.pdb
ligand2_h.pdb → protein2.pdb
ligand3_h.pdb → protein3.pdb
After completion, you should have three separate protein-ligand complex files:
Note: Each file contains exactly one protein structure with its corresponding ligand. Do not combine multiple ligands into a single protein file.
Next Steps
After creating these files, proceed to the parameterization stage as described in the following section.
2. Generating the XYZ Structure with easyPARM
Run easyPARM and select option 2:
Select your option:
1- Generate molecular complex parameters
2- Generate metalloprotein .xyz structure
3- Convert AMBER parameters to OpenMM or GROMACS format
Enter your choice: 2
Please provide the metalloprotein pdb file: protein1.pdb
XYZ Output: initial_structure.xyz
Rename the output file:
mv initial_structure.xyz initial_structure1.xyz
Repeat this for protein2.pdb and protein3.pdb to obtain:
Since protein1.pdb and protein2.pdb produce identical XYZ structures, only one will be used for Optimization, Frequency calculations, and Charge calculations.
3. QM Calculation Outputs
Refer to the QM Input example for guidance on preparing your input files. These examples illustrate how to set up the required calculations for each structure extracted using easyPARM Option 2.
3.1 Protein 1 and Protein 2
- Optimized structure: OPTIMIZED1_2.xyz
- Frequency calculation log: OPT_FREQ1_2.log
- Charge calculation log: CHARGES1_2.log
- Formatted checkpoint file: COMPLEX1_2.fchk
3.2 Protein 3
- Optimized structure: OPTIMIZED3.xyz
- Frequency calculation log: OPT_FREQ3.log
- Charge calculation log: CHARGES3.log
- Formatted checkpoint file: COMPLEX3.fchk
Use this link to download fchk files: fchk
4. Generating Molecular Parameters with easyPARM
Important Notes:
- We will parameterize the ligands one by one, starting with protein 1 and finishing with protein 3.
- It is highly recommended to select the option Change Residue ID to assign a unique residue ID for each heme C.
Run easyPARM again and select option 1:
Enter your choice: 1
This will prompt a series of configurations:
4.1 AMBER Configuration
Choose how to load AMBER:
- Option 1: Use currently loaded AMBER
- Option 2: Specify AMBER installation path
Since AMBER is already loaded, select option 1:
Enter your choice: 1
4.2 System Charge
The tool will request the total charge of the system:
Please provide the total charge: -2
4.3 Providing the Optimized Structure for the Seminario Method
Specify the optimized XYZ geometry file:
Please provide the optimized XYZ geometry file: OPTIMIZED1_2.xyz
4.4 Charge Calculation Method
Choose a method from the menu:
Select the charge calculation method:
1- GAUSSIAN (RESP charges)
2- ORCA (CHELPG charges)
3- ORCA (RESP Charges)
4- GAMESS (RESP Charges)
5- GAMESS (GAMESS Fit Charges)
Enter your choice: 1
4.5 mol2 File Generation
Select the charge output format:
Please select the input format:
1- Gaussian Output (.log file)
2- RESP (.gesp file)
Enter your choice: 1
Select the charge method:
Please select the charge method (recommended: RESP):
1- RESP (resp)
2- Mulliken (mul)
3- ESP (esp)
4- AM1-BCC (bcc)
Enter your choice: 1
Select the atom type:
Please select the atom type:
1- AMBER Force Field (AMBER)
2- General AMBER Force Field (GAFF)
3- General AMBER Force Field (GAFF2)
Enter your choice: 2
Provide the charge output file:
Please provide the charge output file (e.g., .log, .gesp): CHARGES1_2.log
4.6 Seminario Method Setup
Select the format for the Seminario method:
Please select the format you will provide:
1- Orca Output
2- Gaussian Output
3- Gaussian Checkpoint
4- Gaussian Formatted Checkpoint
5- Gamess Output
Enter your choice: 4
Please provide the formatted checkpoint file (.fchk): COMPLEX1_2.fchk
4.7 Metalloprotein Structure Confirmation
Does your structure belong to MetalloProtein? (y/n): y
Provide the final metalloprotein PDB file:
Please provide the metalloprotein PDB file: protein1.pdb
4.8 Change Residue ID
Would you like to change the residue ID (Default= mol)? (y/n): y
Please provide the residue name: 1HE
5. Output Generation
Upon completion, easyPARM generates the following files:
5.1 Key Outputs
- Mol2 files (Residues & Metal):
- Force Field Parameters: COMPLEX_1HE.frcmod
- Library: COMPLEX.lib
- Metalloprotein PDB: easyPARM_MetalloProtein_1HE.pdb
- Bond Information: Bond_Info_1HE.dat
- New Atom Type Definitions: Hybridization_Info_1HE.dat
5.2 Modifications for the Next Step
Copy the PDB output file:
cp easyPARM_MetalloProtein_1HE.pdb temp1.pdb
Open temp1.pdb, locate the first heme with the selected residue ID, and move the metal atom (Fe1) line to a new file:
Next, add ligand2_h.pdb to temp1.pdb. At this stage, we have:
- temp1.pdb: Contains protein, first heme (without metal atom), second heme (with its metal atom).
- reference.pdb: Contains the metal atom (Fe1) of the first heme.
Run easyPARM again and select option 1. Provide the same files (OPTIMIZED1_2.xyz, CHARGES1_2.log, COMPLEX1_2.fchk) but with temp1.pdb and a new residue name.
4.7 Metalloprotein Structure Confirmation (Second Iteration)
Does your structure belong to MetalloProtein? (y/n): y
Provide the final metalloprotein PDB file:
Please provide the metalloprotein PDB file: temp1.pdb
4.8 Change Residue ID
Would you like to change the residue ID (Default= mol)? (y/n): y
Please provide the residue name: 2HE
5. Output Generation
Upon completion, easyPARM generates the following files:
5.1 Key Outputs
- Mol2 files (Residues & Metal):
- Force Field Parameters: COMPLEX_2HE.frcmod
- Library: COMPLEX.lib
- Metalloprotein PDB: easyPARM_MetalloProtein_2HE.pdb
- Bond Information: Bond_Info_2HE.dat
- New Atom Type Definitions: Hybridization_Info_2HE.dat
5.2 Modifications for the Next Step
Copy the PDB output file:
cp easyPARM_MetalloProtein_2HE.pdb temp2.pdb
Modify temp2.pdb by moving the second heme’s metal atom (Fe1) to reference.pdb, then add ligand3_h.pdb. At this stage, we have:
- temp2.pdb: Contains protein, first and second heme (without metal atoms), third heme (with metal atom).
- reference.pdb: Contains metal atoms of the first and second heme.
Run easyPARM again, providing OPTIMIZED3.xyz, CHARGES3.log, COMPLEX3.fchk, and temp2.pdb with a new residue name.
4.7 Metalloprotein Structure Confirmation (Third Iteration)
Does your structure belong to MetalloProtein? (y/n): y
Provide the final metalloprotein PDB file:
Please provide the metalloprotein PDB file: temp2.pdb
4.8 Change Residue ID
Would you like to change the residue ID (Default= mol)? (y/n): y
Please provide the residue name: 3HE
5. Output Generation
Upon completion, easyPARM generates the following files:
5.1 Key Outputs
- Mol2 files (Residues & Metal):
- Force Field Parameters: COMPLEX_3HE.frcmod
- Library: COMPLEX.lib
- Metalloprotein PDB: easyPARM_MetalloProtein_3HE.pdb
- Bond Information: Bond_Info_3HE.dat
- New Atom Type Definitions: Hybridization_Info_3HE.dat
Finalize by merging metal atoms from reference.pdb into the correct residue ID in easyPARM_MetalloProtein_3HE.pdb:
cp easyPARM_MetalloProtein_3HE.pdb easyPARM_MetalloProtein_Final.pdb
This easyPARM_MetalloProtein_Final.pdb final structure is now ready for tleap.
6. Running tleap
Once the parameters are generated, you can proceed with tleap to prepare the final topology and coordinate files. Use the appropriate tleap input file to ensure correct setup:
Streamlined Metalloprotein Library Integration
Our latest implementation now supports the automatic generation of a library file for metalloproteins, eliminating the need for manual handling of multiple parameter files. The generated COMPLEX.lib file simplifies the tleap process by allowing direct loading with:
loadoff COMPLEX.lib
With this approach:
- There is no need to use the
Hybridization_Info.datfile. - You do not need to load each individual
.mol2file manually. - The metalloprotein setup is now more efficient and automated.
tleap Input File
Use the following tleap input file for reference:
- tleap input file: tleap.in
This marks the completion of the metalloprotein parameterization, ensuring a seamless transition to system preparation and simulation.
7. Molecular Dynamics Simulation