![]()
Theoretical Foundation
Most existing parameterization tools primarily target metal complexes or metalloproteins. To address this limitation, easyPARM has been extended to support metal–nucleic acid systems, including:
- Metal-bound DNA and RNA
- Inner-sphere metal coordination to nucleobases or phosphate groups
- Metal-mediated base pairs and metallo-DNA constructs
- Nucleic acid–metal complexes and nucleic acid–directed metal assemblies
The overall workflow closely follows the metalloprotein parameterization strategy described in our published work (DOI: https://doi.org/10.1063/5.0301038), with key modifications specific to DNA and RNA systems.
Key Differences from Metalloprotein Parameterization
-
Capping Strategy In metal–nucleic acid systems, capping atoms are added at the 5′ and 3′ termini of the nucleic acid fragment, rather than at amino acid backbone positions.
- Charge Treatment Unlike metalloprotein capping atoms, which are typically neutral, nucleic acid capping atoms carry a negative charge. Users must therefore take special care when defining the charge of the extracted metal region. For example, a metal complex with a formal charge of +1 may appear neutral after capping. This is expected: the capping atoms are removed at later stages of the workflow, restoring the correct total charge.
- Charge Fixation easyPARM fixes the charges of the DNA or RNA backbone to the standard values defined in AMBER nucleic acid force fields, such as OL21, OL24, and related variants. The backbone treated in this step includes the sugar moiety and the complete phosphate terminus, ensuring full consistency with the selected AMBER force field.
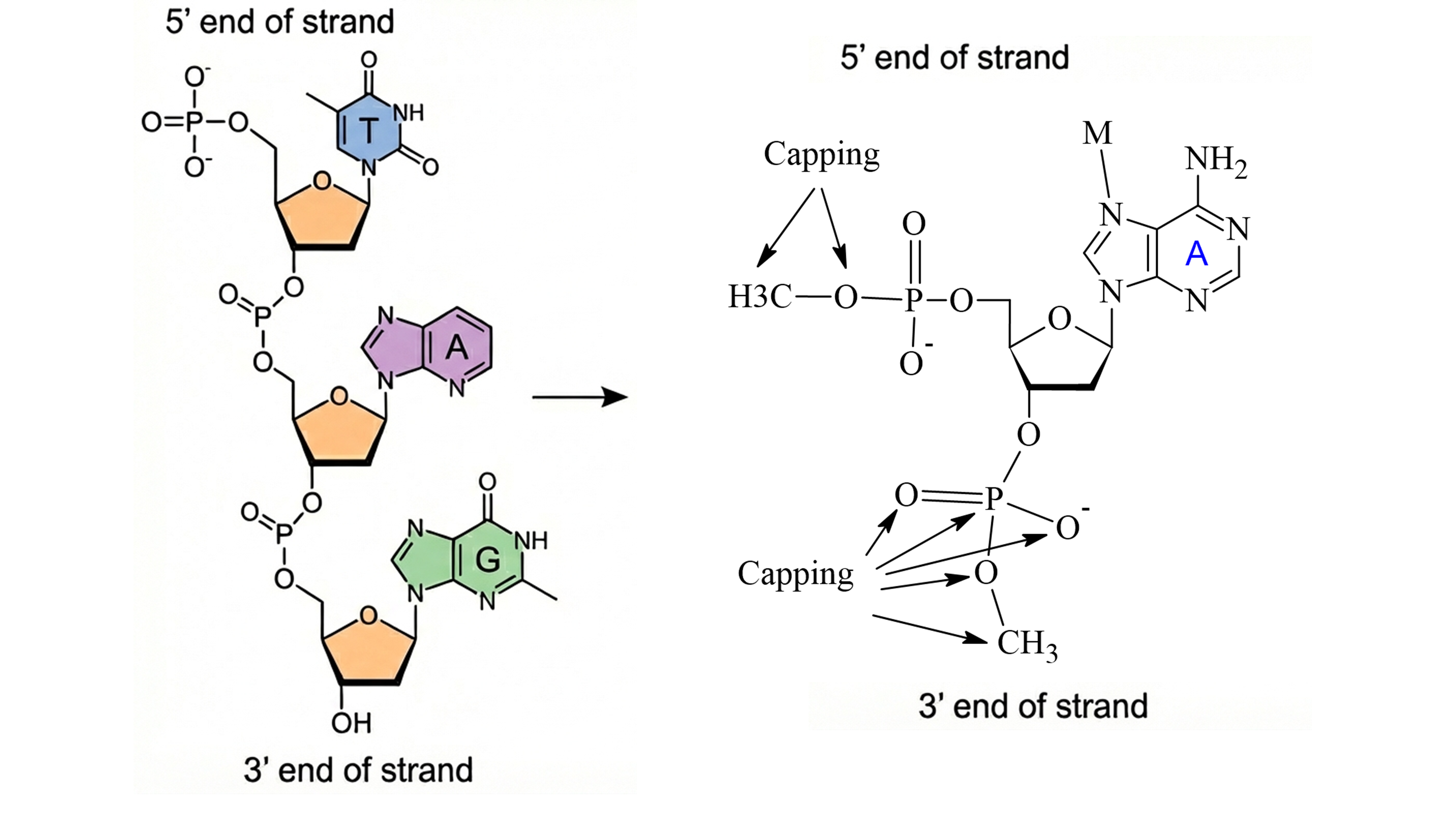
The following sections provide a detailed, step-by-step tutorial illustrating this workflow.
Tutorial: Parameterization of Metal–Nucleic Acid Systems
Overview
This tutorial provides a complete workflow for parameterizing metal–nucleic acid systems using easyPARM in combination with Gaussian for quantum mechanical (QM) calculations. The same procedure can also be applied using ORCA or GAMESS.
As a working example, we use the structure 5BNA.pdb, which contains a DNA fragment coordinated to a platinum atom via the N7 position of guanine.
Note: This tutorial is applicable to any similar system, whether the metal is directly coordinated to the nucleic acid or bound through a metal-containing ligand.
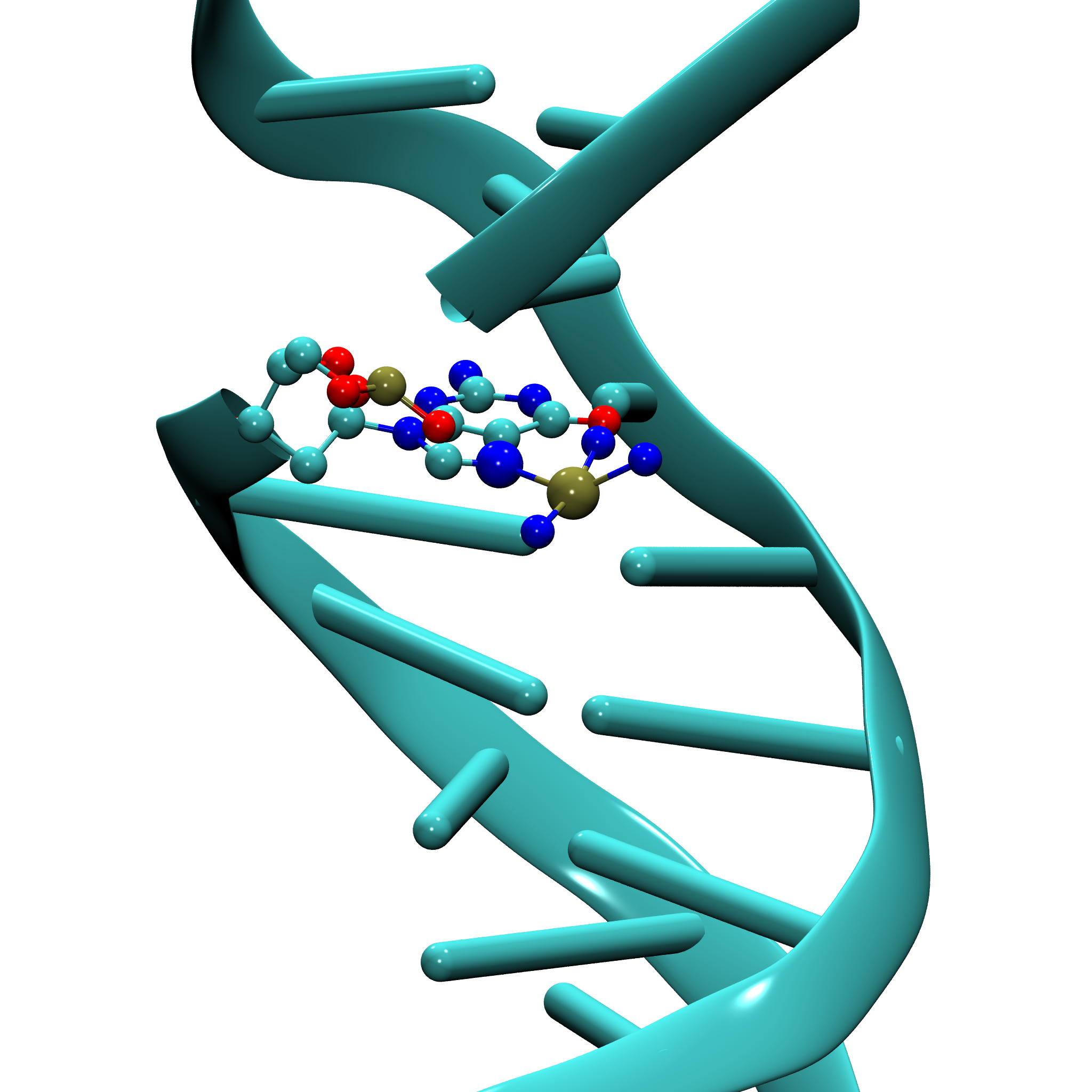
Parameterization Challenges and Strategy
Metal–nucleic acid systems often involve complex coordination environments, non-standard residues, and charge-sensitive fragmentation. This tutorial highlights how easyPARM addresses these challenges and enables reliable force field generation for such systems.
By following this workflow, users can obtain consistent and transferable AMBER-compatible parameters suitable for molecular dynamics simulations of metal–nucleic acid complexes.
Important Steps
To ensure accurate parameterization, each step must be followed carefully.
1. Preprocessing the PDB File
After downloading the structure from the Protein Data Bank, the PDB file must be preprocessed to ensure compatibility with easyPARM and subsequent molecular dynamics simulations.
1.1 Automated Preprocessing
The following tool is used:
Apply H++ to the original 5BNA.pdb. After processing, the output file (e.g., output.pdb) will contain only standard nucleic acid residues. The metal center and any non-standard residues are removed by default.
These components must be manually reintroduced in the next step.
1.2 Manual Preparation
Warning: This step is essential and significantly simplifies the parameterization process.
Step 1: Inspect Metal Coordination
Check whether the metal center is coordinated to non-standard residues or incomplete chemical fragments. If the metal is coordinated exclusively to standard residues, this step may be skipped.
In the present example, platinum is coordinated to three nitrogen atoms without associated hydrogens. Therefore, missing hydrogens must be added.
Use reduce from AmberTools:
reduce -i ligand.pdb -o ligand_H.pdb
- Input: ligand.pdb
- Output: ligand_H.pdb
Finally, merge the corrected non-standard residues and metal center back into the nucleic acid PDB file amber.pdb.
Once this is complete, proceed to the next stage.
2. Generating the XYZ Structure with easyPARM
Run easyPARM and select Option 3:
Select your option:
1- Generate molecular complex parameters
2- Generate metalloprotein .xyz structure
3- Generate metal–nucleic acid system .xyz structure
4- Convert AMBER parameters to OpenMM or GROMACS format
Enter your choice: 3
Then provide the prepared PDB file:
Please provide the metal–nucleic acid system pdb file: amber.pdb
XYZ Output: initial_structure.xyz
The resulting XYZ file serves as the input for QM calculations.
Important: Always visualize the generated
.xyzfile before running QM calculations to confirm that the structure, bonding, and atom ordering are correct.
3. Quantum Mechanical Calculations
Perform geometry optimization, frequency analysis, and charge calculations using your preferred QM software.
3.1 Example QM Outputs (Model 1)
- Gaussian input: OPT_FREQ.com
- Optimized structure: OPTIMIZED.xyz
- Frequency calculation log: OPT_FREQ.log
- Charge calculation log: CHARGES.log
- Formatted checkpoint file: COMPLEX.fchk
4. Generating Force Field Parameters with easyPARM
easyPARM supports both interactive and non-interactive modes. In this tutorial, the non-interactive mode is used.
- General input format: see the non-interactive section
- Example input file: PARAMET.input
In this input file, you must specify:
- Charge calculation output
- Formatted checkpoint file (
.fchk) - Optimized structure
- System type: Metal–Nucleic Acid System
- Original PDB file
- Residue name (e.g., LIG)
5. Output Files
Upon successful completion, easyPARM generates the following outputs.
5.1 Key Outputs
-
Mol2 files (residues and metal):
-
Force field parameters:
-
Library file:
-
Metallo–nucleic acid PDB:
-
Bond information:
-
New atom type definitions:
6. Building the System with tleap
With all parameters prepared, use tleap to generate the topology (.prmtop) and coordinate (.inpcrd) files.
Streamlined Library Integration
The latest version of easyPARM automatically generates a unified library file for metal-containing systems.
Load it directly in tleap:
loadoff COMPLEX.lib
With this approach:
- Manual loading of individual
.mol2files is unnecessary Hybridization_Info.datis not required- System preparation is faster, simpler, and less error-prone
Reference Input
- Example tleap input file: tleap.in
7. Molecular Dynamics Simulation
Once the topology and coordinates are generated, the system is fully prepared for molecular dynamics simulations.
